

文章正文简要报道

蛋白质是具有重要生物学功能的生物大分子,也是许多疾病治疗的靶点。蛋白质由氨基酸通过肽键“串联”形成一维序列(多肽链),这样的一维序列进一步通过分子内相互作用折叠形成特定的三维结构,犹如将魔尺折叠成各种造型的过程。通常,具有一定三维结构的蛋白质才能发挥生物学功能。由于蛋白质结构-功能之间的依赖关系,获取蛋白质的三维结构对研究蛋白质的功能具有重要价值。获取蛋白质三维结构最可靠的途径是利用结构生物学方法,例如X射线衍射、核磁共振、冷冻电镜等,但是这些方法一方面成本(时间、人力、经费)很高,另一方面也都有一定的局限性。例如,X射线衍射对于实验室无法获得晶体的蛋白质无能为力,冷冻电镜对较小的蛋白难以观察。因此,虽然人类目前已知的蛋白质序列有约2亿条,但通过实验获取的蛋白质结构仅有约20万个。利用实验手段解析蛋白质三维结构的困难大大限制了蛋白质功能的研究。

研究内容
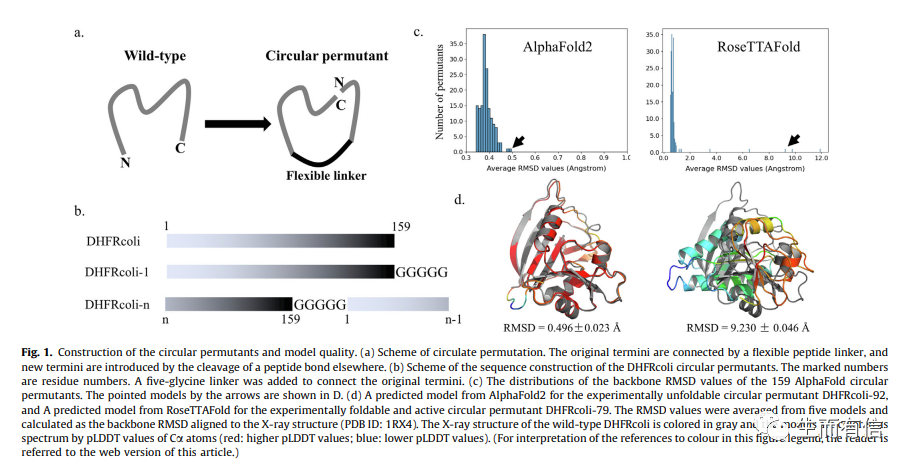
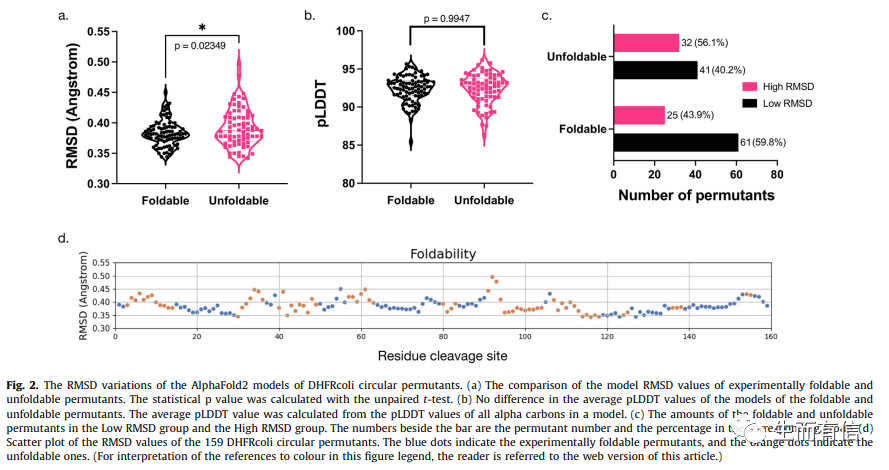
进一步,研究人员设想,AlphaFold2和RoseTTAFold等方法是利用实际可折叠的蛋白质进行学习的,那么是否不可折叠蛋白质序列的“伪三维结构”预测精度会比较差呢?AlphaFold2对预测结构用pLDDT打分评估预测可靠性,然而该打分并不能区分可折叠和不可折叠的DHFR CP突变体。但是研究人员发现可折叠和不可折叠的DHFR CP突变体的预测结构与野生型DHFR晶体结构的差异(RMSD)存在不一样,可折叠的DHFR CP突变体的RMSD更小。进一步研究发现,RMSD的差异可以作为区分可折叠和不可折叠DHFR CP突变体的有效判断标准之一。该发现还在另一套丙氨酸插入产生的DHFR突变体实验数据上得到进一步验证。最后,研究人员还证明这种RMSD差异确实与蛋白的可折叠性而非功能相关。
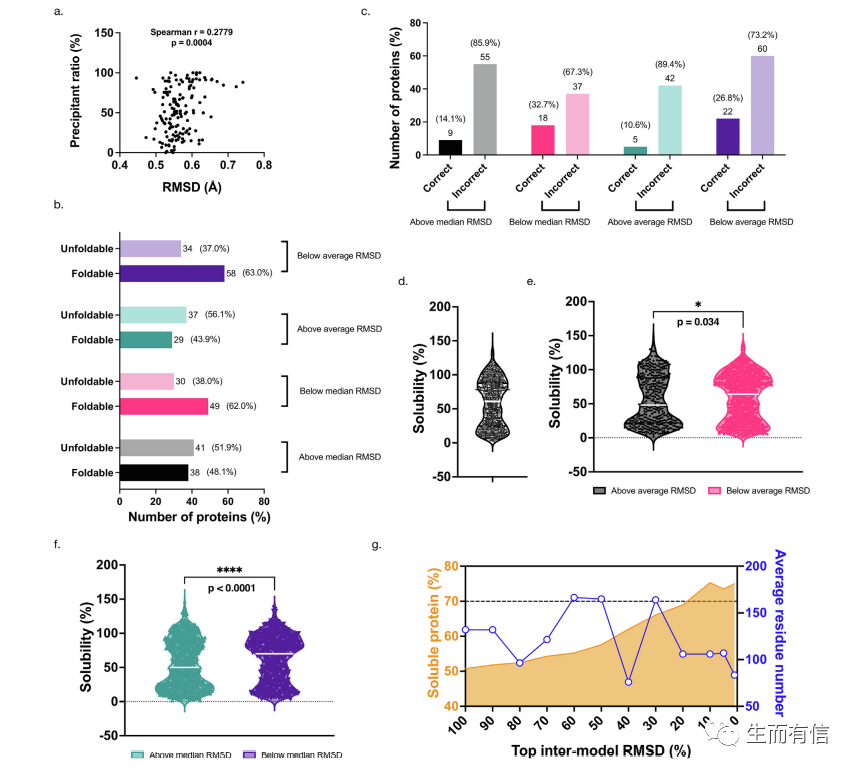
上述RMSD的计算基于有已知结构的天然蛋白质,那么对于没有已知结构的人工设计蛋白质该如何预测其可折叠性呢?研究人员设想,与不可折叠蛋白相比,可折叠蛋白具有更多的序列进化信息,因此不同预测方法得到的结构一致性应该更高。基于这一设想,研究人员利用AlphaFold2和RoseTTAFold两种方法的预测结构间的RMSD差异(inter-model RMSD)分析了DHFR的可折叠性,发现确实存在这样的规律,即可折叠的DHFR序列具有更小的inter-model RMSD。
结语
原文链接:
1. BioRxiv preprint: https://doi.org/10.1101/2022.01.27.477978
2. Computational and Structural Biotechnology Journal: https://doi.org/10.1016/j.csbj.2022.08.034
– 通讯作者简介 –

湖北工业大学
刘森
教授
——— End ———
关于湖北省生物信息学会:
湖北省生物信息学会,是由湖北省内从事生物信息学科技工作者自愿组成的全省性、学术性、非营利性的社会团体。学会致力于制定生物信息学专业规范,加强学术交流与合作,推动人才培养,促进理事单位及省内外生物信息学产业的健康可持续发展。